The RDKit Book¶
Misc Cheminformatics Topics¶
Aromaticity¶
Aromaticity is one of those unpleasant topics that is simultaneously simple and impossibly complicated. Since neither experimental nor theoretical chemists can agree with each other about a definition, it’s necessary to pick something arbitrary and stick to it. This is the approach taken in the RDKit.
Instead of using patterns to match known aromatic systems, the aromaticity perception code in the RDKit uses a set of rules. The rules are relatively straightforward.
Aromaticity is a property of atoms and bonds in rings. An aromatic bond must be between aromatic atoms, but a bond between aromatic atoms does not need to be aromatic.
For example the fusing bonds here are not considered to be aromatic by the RDKit:
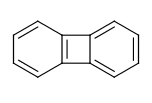
>>> from rdkit import Chem
>>> m = Chem.MolFromSmiles('C1=CC2=C(C=C1)C1=CC=CC=C21')
>>> m.GetAtomWithIdx(3).GetIsAromatic()
True
>>> m.GetAtomWithIdx(6).GetIsAromatic()
True
>>> m.GetBondBetweenAtoms(3,6).GetIsAromatic()
False
The RDKit supports a number of different aromaticity models and allows the user to define their own by providing a function that assigns aromaticity.
The RDKit Aromaticity Model¶
A ring, or fused ring system, is considered to be aromatic if it obeys the 4N+2 rule. Contributions to the electron count are determined by atom type and environment. Some examples:
| Fragment | Number of pi electrons |
| c(a)a | 1 |
| n(a)a | 1 |
| An(a)a | 2 |
| o(a)a | 2 |
| s(a)a | 2 |
| se(a)a | 2 |
| te(a)a | 2 |
| O=c(a)a | 0 |
| N=c(a)a | 0 |
| *(a)a | 0, 1, or 2 |
Notation a: any aromatic atom; A: any atom, include H; *: a dummy atom
Notice that exocyclic bonds to electronegative atoms “steal” the valence electron from the ring atom and that dummy atoms contribute whatever count is necessary to make the ring aromatic.
The use of fused rings for aromaticity can lead to situations where individual rings are not aromatic, but the fused system is. An example of this is azulene:
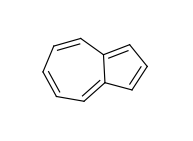
An extreme example, demonstrating both fused rings and the influence of exocyclic double bonds:
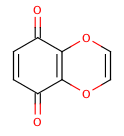
>>> m=Chem.MolFromSmiles('O=C1C=CC(=O)C2=C1OC=CO2')
>>> m.GetAtomWithIdx(6).GetIsAromatic()
True
>>> m.GetAtomWithIdx(7).GetIsAromatic()
True
>>> m.GetBondBetweenAtoms(6,7).GetIsAromatic()
False
A special case, heteroatoms with radicals are not considered candidates for aromaticity:
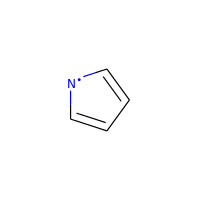
>>> m = Chem.MolFromSmiles('C1=C[N]C=C1')
>>> m.GetAtomWithIdx(0).GetIsAromatic()
False
>>> m.GetAtomWithIdx(2).GetIsAromatic()
False
>>> m.GetAtomWithIdx(2).GetNumRadicalElectrons()
1
Charged carbons with radicals are also not considered:
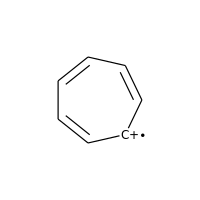
>>> m = Chem.MolFromSmiles('C1=CC=CC=C[C+]1')
>>> m.GetAtomWithIdx(0).GetIsAromatic()
False
>>> m.GetAtomWithIdx(6).GetIsAromatic()
False
>>> m.GetAtomWithIdx(6).GetFormalCharge()
1
>>> m.GetAtomWithIdx(6).GetNumRadicalElectrons()
1
Neutral carbons with radicals, however, are still considered:
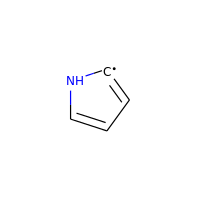
>>> m = Chem.MolFromSmiles('C1=[C]NC=C1')
>>> m.GetAtomWithIdx(0).GetIsAromatic()
True
>>> m.GetAtomWithIdx(1).GetIsAromatic()
True
>>> m.GetAtomWithIdx(1).GetNumRadicalElectrons()
1
The Simple Aromaticity Model¶
This one is quite simple: only five- and six-membered simple rings are considered candidates for aromaticity. The same electron-contribution counts listed above are used.
The MDL Aromaticity Model¶
This isn’t well documented (at least not publicly), so we tried to reproduce what’s provided in the oechem documentation (https://docs.eyesopen.com/toolkits/python/oechemtk/aromaticity.html)
- fused rings (i.e. azulene) can be aromatic
- five-membered rings are not aromatic (though they can be part of fused aromatic systems)
- only C and N can be aromatic
- only one electron donors are accepted
- atoms with exocyclic double bonds are not aromatic
Note: For reasons of computational expediency, aromaticity perception is only done for fused-ring systems where all members are at most 24 atoms in size.
SMILES Support and Extensions¶
The RDKit covers all of the standard features of Daylight SMILES [2] as well as some useful extensions.
Here’s the (likely partial) list of extensions:
Aromaticity¶
te(aromatic Te) is accepted
Dative bonds¶
<- and -> create a dative bond between the atoms, direction does matter.
Here’s an example of a bipy-copper complex:
>>> bipycu = Chem.MolFromSmiles('c1cccn->2c1-c1n->3cccc1.[Pt]23(Cl)Cl')
>>> bipycu.GetBondBetweenAtoms(4,12).GetBondType()
rdkit.Chem.rdchem.BondType.DATIVE
>>> Chem.MolToSmiles(bipycu)
'Cl[Pt]1(Cl)<-n2ccccc2-c2ccccn->12'
Dative bonds have the special characteristic that they don’t affect the valence on the start atom, but do affect the end atom. So in this case, the N atoms involved in the dative bond have the valence of 3 that we expect from bipy, while the Cu has a valence of 4:
>>> bipycu.GetAtomWithIdx(4).GetTotalValence()
3
>>> bipycu.GetAtomWithIdx(12).GetTotalValence()
4
Specifying atoms by atomic number¶
The [#6] construct from SMARTS is supported in SMILES.
CXSMILES extensions¶
The RDKit supports parsing and writing a subset of the extended SMILES functionality introduced by ChemAxon [#cxsmiles]_CIPCode
The features which are parsed include:
- atomic coordinates
- atomic values
- atomic labels
- atomic properties
- coordinate bonds (these are translated into double bonds)
- radicals
- enhanced stereo (these are converted into
StereoGroups)
The features which are written by rdkit.Chem.rdmolfiles.MolToCXSmiles()
(note the specialized writer function) include:
- atomic coordinates
- atomic values
- atomic labels
- atomic properties
- radicals
- enhanced stereo
>>> m = Chem.MolFromSmiles('OC')
>>> m.GetAtomWithIdx(0).SetProp('p1','2')
>>> m.GetAtomWithIdx(1).SetProp('p1','5')
>>> m.GetAtomWithIdx(1).SetProp('p2','A1')
>>> m.GetAtomWithIdx(0).SetProp('atomLabel','O1')
>>> m.GetAtomWithIdx(1).SetProp('atomLabel','C2')
>>> Chem.MolToCXSmiles(m)
'CO |$C2;O1$,atomProp:0.p1.5:0.p2.A1:1.p1.2|'
SMARTS Support and Extensions¶
The RDKit covers most of the standard features of Daylight SMARTS [3] as well as some useful extensions.
Here’s the (hopefully complete) list of SMARTS features that are not supported:
- Non-tetrahedral chiral classes
- the
@?operator - explicit atomic masses (though isotope queries are supported)
- component level grouping requiring matches in different components, i.e.
(C).(C)
Here’s the (likely partial) list of extensions:
Hybridization queries¶
^0matches S hybridized atoms^1matches SP hybridized atoms^2matches SP2 hybridized atoms^3matches SP3 hybridized atoms^4matches SP3D hybridized atoms^5matches SP3D2 hybridized atoms
>> Chem.MolFromSmiles('CC=CF').GetSubstructMatches(Chem.MolFromSmarts('[^2]'))
((1,), (2,))
Dative bonds¶
<- and -> match the corresponding dative bonds, direction does matter.
>>> Chem.MolFromSmiles('C1=CC=CC=N1->[Fe]').GetSubstructMatches(Chem.MolFromSmarts('[#7]->*'))
((5, 6),)
>>> Chem.MolFromSmiles('C1=CC=CC=N1->[Fe]').GetSubstructMatches(Chem.MolFromSmarts('*<-[#7]'))
((6, 5),)
Heteroatom neighbor queries¶
- the atom query
zmatches atoms that have the specified number of heteroatom (i.e. not C or H) neighbors. For example,z2would match the second C inCC(=O)O.- the atom query
Zmatches atoms that have the specified number of aliphatic heteroatom (i.e. not C or H) neighbors.
>>> Chem.MolFromSmiles('O=C(O)c1nc(O)ccn1').GetSubstructMatches(Chem.MolFromSmarts('[z2]'))
((1,), (3,), (5,))
>>> Chem.MolFromSmiles('O=C(O)c1nc(O)ccn1').GetSubstructMatches(Chem.MolFromSmarts('[Z2]'))
((1,),)
>>> Chem.MolFromSmiles('O=C(O)c1nc(O)ccn1').GetSubstructMatches(Chem.MolFromSmarts('[Z1]'))
((5,),)
Range queries¶
Ranges of values can be provided for many query types that expect numeric values.
The query types that currently support range queries are:
D, h, r, R, v, x, X, z, Z
- Here are some examples:
D{2-4}matches atoms that have between 2 and 4 (inclusive) explicit connections.D{-3}matches atoms that have less than or equal to 3 explicit connections.D{2-}matches atoms that have at least 2 explicit connections.
>>> Chem.MolFromSmiles('CC(=O)OC').GetSubstructMatches(Chem.MolFromSmarts('[z{1-}]'))
((1,), (4,))
>>> Chem.MolFromSmiles('CC(=O)OC').GetSubstructMatches(Chem.MolFromSmarts('[D{2-3}]'))
((1,), (3,))
>>> Chem.MolFromSmiles('CC(=O)OC.C').GetSubstructMatches(Chem.MolFromSmarts('[D{-2}]'))
((0,), (2,), (3,), (4,), (5,))
Ring Finding and SSSR¶
[Section taken from “Getting Started” document]
As others have ranted about with more energy and eloquence than I intend to, the definition of a molecule’s smallest set of smallest rings is not unique. In some high symmetry molecules, a “true” SSSR will give results that are unappealing. For example, the SSSR for cubane only contains 5 rings, even though there are “obviously” 6. This problem can be fixed by implementing a small (instead of smallest) set of smallest rings algorithm that returns symmetric results. This is the approach that we took with the RDKit.
Because it is sometimes useful to be able to count how many SSSR rings are present in the molecule, there is a GetSSSR function, but this only returns the SSSR count, not the potentially non-unique set of rings.
For situations where you just care about knowing whether or not atoms/bonds are in rings, the RDKit provides the function
rdkit.Chem.rdmolops.FastFindRings(). This does a depth-first traversal of the molecule graph and identifies atoms and bonds that
are in rings.
Chemical Reaction Handling¶
Reaction SMARTS¶
Not SMIRKS [1] , not reaction SMILES [2], derived from SMARTS [3].
The general grammar for a reaction SMARTS is :
reaction ::= reactants ">>" products
reactants ::= molecules
products ::= molecules
molecules ::= molecule
molecules "." molecule
molecule ::= a valid SMARTS string without "." characters
"(" a valid SMARTS string without "." characters ")"
Some features¶
Mapped dummy atoms in the product template are replaced by the corresponding atom in the reactant:
>>> from rdkit.Chem import AllChem
>>> rxn = AllChem.ReactionFromSmarts('[C:1]=[O,N:2]>>[C:1][*:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('CC=O'),))[0]]
['CCO']
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('CC=N'),))[0]]
['CCN']
but unmapped dummy atoms are left as dummies:
>>> rxn = AllChem.ReactionFromSmarts('[C:1]=[O,N:2]>>*[C:1][*:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('CC=O'),))[0]]
['*C(C)O']
“Any” bonds in the products are replaced by the corresponding bond in the reactant:
>>> rxn = AllChem.ReactionFromSmarts('[C:1]~[O,N:2]>>*[C:1]~[*:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('C=O'),))[0]]
['*C=O']
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('CO'),))[0]]
['*CO']
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('C#N'),))[0]]
['*C#N']
Intramolecular reactions can be expressed flexibly by including reactants in parentheses. This is demonstrated in this ring-closing metathesis example [5]:
>>> rxn = AllChem.ReactionFromSmarts("([C:1]=[C;H2].[C:2]=[C;H2])>>[*:1]=[*:2]")
>>> m1 = Chem.MolFromSmiles('C=CCOCC=C')
>>> ps = rxn.RunReactants((m1,))
>>> Chem.MolToSmiles(ps[0][0])
'C1=CCOC1'
Chirality¶
This section describes how chirality information in the reaction defition is handled. A consistent example, esterification of secondary alcohols, is used throughout [6].
If no chiral information is present in the reaction definition, the stereochemistry of the reactants is preserved, as is membership in enhanced stereo groups:
>>> alcohol1 = Chem.MolFromSmiles('CC(CCN)O')
>>> alcohol2 = Chem.MolFromSmiles('C[C@H](CCN)O')
>>> alcohol3 = Chem.MolFromSmiles('C[C@@H](CCN)O')
>>> acid = Chem.MolFromSmiles('CC(=O)O')
>>> rxn = AllChem.ReactionFromSmarts('[CH1:1][OH:2].[OH][C:3]=[O:4]>>[C:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@@H](C)CCN'
You get the same result (retention of stereochemistry) if a mapped atom has the same chirality in both reactants and products:
>>> rxn = AllChem.ReactionFromSmarts('[C@H1:1][OH:2].[OH][C:3]=[O:4]>>[C@:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@@H](C)CCN'
A mapped atom with different chirality in reactants and products leads to inversion of stereochemistry:
>>> rxn = AllChem.ReactionFromSmarts('[C@H1:1][OH:2].[OH][C:3]=[O:4]>>[C@@:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
If a mapped atom has chirality specified in the reactants, but not in the products, the reaction destroys chirality at that center:
>>> rxn = AllChem.ReactionFromSmarts('[C@H1:1][OH:2].[OH][C:3]=[O:4]>>[C:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)OC(C)CCN'
And, finally, if chirality is specified in the products, but not the reactants, the reaction creates a stereocenter with the specified chirality:
>>> rxn = AllChem.ReactionFromSmarts('[CH1:1][OH:2].[OH][C:3]=[O:4]>>[C@:1][O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
Note that this doesn’t make sense without including a bit more context around the stereocenter in the reaction definition:
>>> rxn = AllChem.ReactionFromSmarts('[CH3:5][CH1:1]([C:6])[OH:2].[OH][C:3]=[O:4]>>[C:5][C@:1]([C:6])[O:2][C:3]=[O:4]')
>>> ps=rxn.RunReactants((alcohol1,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol2,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
>>> ps=rxn.RunReactants((alcohol3,acid))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(=O)O[C@H](C)CCN'
Note that the chirality specification is not being used as part of the query: a molecule with no chirality specified can match a reactant with specified chirality.
In general, the reaction machinery tries to preserve as much stereochemistry information as possible. This works when a single new bond is formed to a chiral center:
>>> rxn = AllChem.ReactionFromSmarts('[C:1][C:2]-O>>[C:1][C:2]-S')
>>> alcohol2 = Chem.MolFromSmiles('C[C@@H](O)CCN')
>>> ps=rxn.RunReactants((alcohol2,))
>>> Chem.MolToSmiles(ps[0][0],True)
'C[C@@H](S)CCN'
But it fails if two or more bonds are formed:
>>> rxn = AllChem.ReactionFromSmarts('[C:1][C:2](-O)-F>>[C:1][C:2](-S)-Cl')
>>> alcohol = Chem.MolFromSmiles('C[C@@H](O)F')
>>> ps=rxn.RunReactants((alcohol,))
>>> Chem.MolToSmiles(ps[0][0],True)
'CC(S)Cl'
In this case, there’s just not sufficient information present to allow the information to be preserved. You can help by providing mapping information:
Rules and caveats¶
- Include atom map information at the end of an atom query. So do [C,N,O:1] or [C;R:1].
- Don’t forget that unspecified bonds in SMARTS are either single or aromatic. Bond orders in product templates are assigned when the product template itself is constructed and it’s not always possible to tell if the bond should be single or aromatic:
>>> rxn = AllChem.ReactionFromSmarts('[#6:1][#7,#8:2]>>[#6:1][#6:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('C1NCCCC1'),))[0]]
['C1CCCCC1']
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('c1ncccc1'),))[0]]
['c1ccccc-1']
So if you want to copy the bond order from the reactant, use an “Any” bond:
>>> rxn = AllChem.ReactionFromSmarts('[#6:1][#7,#8:2]>>[#6:1]~[#6:2]')
>>> [Chem.MolToSmiles(x,1) for x in rxn.RunReactants((Chem.MolFromSmiles('c1ncccc1'),))[0]]
['c1ccccc1']
The Feature Definition File Format¶
An FDef file contains all the information needed to define a set of chemical features. It contains definitions of feature types that are defined from queries built up using Daylight’s SMARTS language. [3] The FDef file can optionally also include definitions of atom types that are used to make feature definitions more readable.
Chemical Features¶
Chemical features are defined by a Feature Type and a Feature Family. The Feature Family is a general classification of the feature (such as “Hydrogen-bond Donor” or “Aromatic”) while the Feature Type provides additional, higher-resolution, information about features. Pharmacophore matching is done using Feature Family’s. Each feature type contains the following pieces of information:
- A SMARTS pattern that describes atoms (one or more) matching the feature type.
- Weights used to determine the feature’s position based on the positions of its defining atoms.
Syntax of the FDef file¶
AtomType definitions¶
An AtomType definition allows you to assign a shorthand name to be used in place of a SMARTS string defining an atom query. This allows FDef files to be made much more readable. For example, defining a non-polar carbon atom like this:
AtomType Carbon_NonPolar [C&!$(C=[O,N,P,S])&!$(C#N)]
creates a new name that can be used anywhere else in the FDef file that it would be useful to use this SMARTS. To reference an AtomType, just include its name in curly brackets. For example, this excerpt from an FDef file defines another atom type - Hphobe - which references the Carbon_NonPolar definition:
AtomType Carbon_NonPolar [C&!$(C=[O,N,P,S])&!$(C#N)]
AtomType Hphobe [{Carbon_NonPolar},c,s,S&H0&v2,F,Cl,Br,I]
Note that {Carbon_NonPolar} is used in the new AtomType definition without any additional decoration (no square brackes or recursive SMARTS markers are required).
Repeating an AtomType results in the two definitions being combined using the SMARTS “,” (or) operator. Here’s an example:
AtomType d1 [N&!H0]
AtomType d1 [O&!H0]
This is equivalent to:
AtomType d1 [N&!H0,O&!H0]
Which is equivalent to the more efficient:
AtomType d1 [N,O;!H0]
Note that these examples tend to use SMARTS’s high-precendence and operator “&” and not the low-precedence and “;”. This can be important when AtomTypes are combined or when they are repeated. The SMARTS “,” operator is higher precedence than “;”, so definitions that use “;” can lead to unexpected results.
It is also possible to define negative AtomType queries:
AtomType d1 [N,O,S]
AtomType !d1 [H0]
The negative query gets combined with the first to produce a definition identical to this:
AtomType d1 [!H0;N,O,S]
Note that the negative AtomType is added to the beginning of the query.
Feature definitions¶
A feature definition is more complex than an AtomType definition and stretches across multiple lines:
DefineFeature HDonor1 [N,O;!H0]
Family HBondDonor
Weights 1.0
EndFeature
The first line of the feature definition includes the feature type and the SMARTS string defining the feature. The next two lines (order not important) define the feature’s family and its atom weights (a comma-delimited list that is the same length as the number of atoms defining the feature). The atom weights are used to calculate the feature’s locations based on a weighted average of the positions of the atom defining the feature. More detail on this is provided below. The final line of a feature definition must be EndFeature. It is perfectly legal to mix AtomType definitions with feature definitions in the FDef file. The one rule is that AtomTypes must be defined before they are referenced.
Additional syntax notes:¶
Any line that begins with a # symbol is considered a comment and will be ignored.
A backslash character, , at the end of a line is a continuation character, it indicates that the data from that line is continued on the next line of the file. Blank space at the beginning of these additional lines is ignored. For example, this AtomType definition:
AtomType tButylAtom [$([C;!R](-[CH3])(-[CH3])(-[CH3])),\ $([CH3](-[C;!R](-[CH3])(-[CH3])))]
is exactly equivalent to this one:
AtomType tButylAtom [$([C;!R](-[CH3])(-[CH3])(-[CH3])),$([CH3](-[C;!R](-[CH3])(-[CH3])))]
(though the first form is much easier to read!)
Atom weights and feature locations¶
Frequently Asked Question(s)¶
What happens if a Feature Type is repeated in the file? Here’s an example:
DefineFeature HDonor1 [O&!H0] Family HBondDonor Weights 1.0 EndFeature DefineFeature HDonor1 [N&!H0] Family HBondDonor Weights 1.0 EndFeature
In this case both definitions of the HDonor1 feature type will be active. This is functionally identical to:
DefineFeature HDonor1 [O,N;!H0] Family HBondDonor Weights 1.0 EndFeature
However the formulation of this feature definition with a duplicated feature type is considerably less efficient and more confusing than the simpler combined definition.
Representation of Pharmacophore Fingerprints¶
In the RDKit scheme the bit ids in pharmacophore fingerprints are not hashed: each bit corresponds to a particular combination of features and distances. A given bit id can be converted back to the corresponding feature types and distances to allow interpretation. An illustration for 2D pharmacophores is shown in Figure 1: Bit numbering in pharmacophore fingerprints.

Figure 1: Bit numbering in pharmacophore fingerprints
Atom-Atom Matching in Substructure Queries¶
When doing substructure matches for queries derived from SMARTS the rules for which atoms in the molecule should match which atoms in the query are well defined.[#smarts]_ The same is not necessarily the case when the query molecule is derived from a mol block or SMILES.
The general rule used in the RDKit is that if you don’t specify a property in the query, then it’s not used as part of the matching criteria and that Hs are ignored. This leads to the following behavior:
| Molecule | Query | Match |
|---|---|---|
| CCO | CCO | Yes |
| CC[O-] | CCO | Yes |
| CCO | CC[O-] | No |
| CC[O-] | CC[O-] | Yes |
| CC[O-] | CC[OH] | Yes |
| CCOC | CC[OH] | Yes |
| CCOC | CCO | Yes |
| CCC | CCC | Yes |
| CC[14C] | CCC | Yes |
| CCC | CC[14C] | No |
| CC[14C] | CC[14C] | Yes |
| OCO | C | Yes |
| OCO | [CH] | No |
| OCO | [CH2] | No |
| OCO | [CH3] | No |
| OCO | O[CH3] | Yes |
| O[CH2]O | C | Yes |
| O[CH2]O | [CH2] | No |
Demonstrated here:
>>> Chem.MolFromSmiles('CCO').HasSubstructMatch(Chem.MolFromSmiles('CCO'))
True
>>> Chem.MolFromSmiles('CC[O-]').HasSubstructMatch(Chem.MolFromSmiles('CCO'))
True
>>> Chem.MolFromSmiles('CCO').HasSubstructMatch(Chem.MolFromSmiles('CC[O-]'))
False
>>> Chem.MolFromSmiles('CC[O-]').HasSubstructMatch(Chem.MolFromSmiles('CC[O-]'))
True
>>> Chem.MolFromSmiles('CC[O-]').HasSubstructMatch(Chem.MolFromSmiles('CC[OH]'))
True
>>> Chem.MolFromSmiles('CCOC').HasSubstructMatch(Chem.MolFromSmiles('CC[OH]'))
True
>>> Chem.MolFromSmiles('CCOC').HasSubstructMatch(Chem.MolFromSmiles('CCO'))
True
>>> Chem.MolFromSmiles('CCC').HasSubstructMatch(Chem.MolFromSmiles('CCC'))
True
>>> Chem.MolFromSmiles('CC[14C]').HasSubstructMatch(Chem.MolFromSmiles('CCC'))
True
>>> Chem.MolFromSmiles('CCC').HasSubstructMatch(Chem.MolFromSmiles('CC[14C]'))
False
>>> Chem.MolFromSmiles('CC[14C]').HasSubstructMatch(Chem.MolFromSmiles('CC[14C]'))
True
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('C'))
True
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('[CH]'))
False
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('[CH2]'))
False
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('[CH3]'))
False
>>> Chem.MolFromSmiles('OCO').HasSubstructMatch(Chem.MolFromSmiles('O[CH3]'))
True
>>> Chem.MolFromSmiles('O[CH2]O').HasSubstructMatch(Chem.MolFromSmiles('C'))
True
>>> Chem.MolFromSmiles('O[CH2]O').HasSubstructMatch(Chem.MolFromSmiles('[CH2]'))
False
Molecular Sanitization¶
The molecule parsing functions all, by default, perform a “sanitization” operation on the molecules read. The idea is to generate useful computed properties (like hybridization, ring membership, etc.) for the rest of the code and to ensure that the molecules are “reasonable”: that they can be represented with octet-complete Lewis dot structures.
Here are the steps involved, in order.
clearComputedProps: removes any computed properties that already existon the molecule and its atoms and bonds. This step is always performed.
cleanUp: standardizes a small number of non-standard valence states. The clean up operations are:
- Neutral 5 valent Ns with double bonds to Os are converted to the zwitterionic form. Example:
N(=O)=O -> [N+](=O)O-]- Neutral 5 valent Ns with triple bonds to another N are converted to the zwitterionic form. Example:
C-N=N#N -> C-N=[N+]=[N-]- Neutral 5 valent phosphorus with one double bond to an O and another to either a C or a P are converted to the zwitterionic form. Example:
C=P(=O)O -> C=[P+]([O-])O- Neutral Cl, Br, or I with exclusively O neighbors, and a valence of 3, 5, or 7, are converted to the zwitterionic form. This covers things like chlorous acid, chloric acid, and perchloric acid. Example:
O=Cl(=O)O -> [O-][Cl+2][O-]OThis step should not generate execptions.
updatePropertyCache: calculates the explicit and implicit valences on all atoms. This generates exceptions for atoms in higher-than-allowed valence states. This step is always performed, but if it is “skipped” the test for non-standard valences will not be carried out.
symmetrizeSSSR: calls the symmetrized smallest set of smallest rings algorithm (discussed in the Getting Started document).
Kekulize: converts aromatic rings to their Kekule form. Will raise an exception if a ring cannot be kekulized or if aromatic bonds are found outside of rings.
assignRadicals: determines the number of radical electrons (if any) on each atom.
setAromaticity: identifies the aromatic rings and ring systems (see above), sets the aromatic flag on atoms and bonds, sets bond orders to aromatic.
setConjugation: identifies which bonds are conjugated
setHybridization: calculates the hybridization state of each atom
cleanupChirality: removes chiral tags from atoms that are not sp3 hybridized.
adjustHs: adds explicit Hs where necessary to preserve the chemistry. This is typically needed for heteroatoms in aromatic rings. The classic example is the nitrogen atom in pyrrole.
The individual steps can be toggled on or off when calling
MolOps::sanitizeMol or Chem.SanitizeMol.
Implementation Details¶
“Magic” Property Values¶
The following property values are regularly used in the RDKit codebase and may be useful to client code.
ROMol (Mol in Python)¶
| Property Name | Use |
|---|---|
| MolFileComments | Read from/written to the comment line of CTABs. |
| MolFileInfo | Read from/written to the info line of CTABs. |
| _MolFileChiralFlag | Read from/written to the chiral flag of CTABs. |
| _Name | Read from/written to the name line of CTABs. |
| _smilesAtomOutputOrder | The order in which atoms were written to SMILES |
Atom¶
| Property Name | Use |
|---|---|
| _CIPCode | the CIP code (R or S) of the atom |
| _CIPRank | the integer CIP rank of the atom |
| _ChiralityPossible | set if an atom is a possible chiral center |
| _MolFileRLabel | integer R group label for an atom, read from/written to CTABs. |
| _ReactionDegreeChanged | set on an atom in a product template of a reaction if its degree changes in the reaction |
| _protected | atoms with this property set will not be considered as matching reactant queries in reactions |
| dummyLabel | (on dummy atoms) read from/written to CTABs as the atom symbol |
| molAtomMapNumber | the atom map number for an atom, read from/written to SMILES and CTABs |
| molfileAlias | the mol file alias for an atom (follows A tags), read from/written to CTABs |
| molFileValue | the mol file value for an atom (follows V tags), read from/written to CTABs |
| molFileInversionFlag | used to flag whether stereochemistry at an atom changes in a reaction, read from/written to CTABs, determined automatically from SMILES |
| molRxnComponent | which component of a reaction an atom belongs to, read from/written to CTABs |
| molRxnRole | which role an atom plays in a reaction (1=Reactant, 2=Product, 3=Agent), read from/written to CTABs |
| smilesSymbol | determines the symbol that will be written to a SMILES for the atom |
Thread safety and the RDKit¶
While writing the RDKit, we did attempt to ensure that the code would work in a multi-threaded environment by avoiding use of global variables, etc. However, making code thread safe is not a completely trivial thing, so there are no doubt some gaps. This section describes which pieces of the code base have explicitly been tested for thread safety.
- Note: With the exception of the small number of methods/functions
- that take a
numThreadsargument, this section does not apply to using the RDKit from Python threads. Boost.Python ensures that only one thread is calling into the C++ code at any point. To get concurrent execution in Python, use the multiprocessing module or one of the other standard python approaches for this .
What has been tested¶
- Reading molecules from SMILES/SMARTS/Mol blocks
- Writing molecules to SMILES/SMARTS/Mol blocks
- Generating 2D coordinates
- Generating 3D conformations with the distance geometry code
- Optimizing molecules with UFF or MMFF
- Generating fingerprints
- The descriptor calculators in $RDBASE/Code/GraphMol/Descriptors
- Substructure searching (Note: if a query molecule contains recursive queries, it may not be safe to use it concurrently on multiple threads, see below)
- The Subgraph code
- The ChemTransforms code
- The chemical reactions code
- The Open3DAlign code
- The MolDraw2D drawing code
Known Problems¶
- InChI generation and (probably) parsing. This seems to be a limitation of the IUPAC InChI code. In order to allow the code to be used in a multi-threaded environment, a mutex is used to ensure that only one thread is using the IUPAC code at a time. This is only enabled if the RDKit is built with the
RDK_TEST_MULTITHREADEDoption enabled.- The MolSuppliers (e.g. SDMolSupplier, SmilesMolSupplier?) change their internal state when a molecule is read. It is not safe to use one supplier on more than one thread.
- Substructure searching using query molecules that include recursive queries. The recursive queries modify their internal state when a search is run, so it’s not safe to use the same query concurrently on multiple threads. If the code is built using the
RDK_BUILD_THREADSAFE_SSSargument (the default for the binaries we provide), a mutex is used to ensure that only one thread is using a given recursive query at a time.
Implementation of the TPSA Descriptor¶
The topological polar surface area (TPSA) descriptor implemented in the RDKit is described in a publication by Peter Ertl et al. (https://pubs.acs.org/doi/abs/10.1021/jm000942e) The RDKit’s implementation differs from what is described in that publication. This section describes the difference and why it’s there.
The RDKit’s TPSA implementation only includes, by default, contributions from N and O atoms. Table 1 of the TPSA publication. however, includes parameters for polar S and P in addition to N and O. What’s going on?
The original TPSA implementation that is in the Daylight Contrib dir (http://www.daylight.com/download/contrib/tpsa.html) does not include contributions from polar S or P and, it turns out, the reference values that are included in the TPSA paper also don’t include S or P contributions. For example, the TPSA provided in Table 3 for foscarnet (SMILES OC(=O)P(=O)(O)O), 94.8, corresponds the sum of the O contributions - 3x20.23 + 2*17.07 = 94.8. Adding the P contribution - 9.81- would give a PSA of 104.6. This is also true for the other P and S containing compounds in Table 3.
In the RDKit implementation, we chose to reproduce the behavior of the tpsa.c Contrib program and what is provided in Table 3 of the paper, so polar S and P are ignored. Based on a couple of user requests, for the 2018.09 release of the RDKit we added the option to include S and P contributions:
>>> from rdkit.Chem import Descriptors
>>> Descriptors.TPSA(Chem.MolFromSmiles('OC(=O)P(=O)(O)O')) # foscarnet
94.83
>>> Descriptors.TPSA(Chem.MolFromSmiles('OC(=O)P(=O)(O)O'), includeSandP=True)
104.64...
>>> Descriptors.TPSA(Chem.MolFromSmiles('Cc1ccccc1N1C(=O)c2cc(S(N)(=O)=O)c(Cl)cc2NC1C')) # metolazone
92.5
>>> Descriptors.TPSA(Chem.MolFromSmiles('Cc1ccccc1N1C(=O)c2cc(S(N)(=O)=O)c(Cl)cc2NC1C'), includeSandP=True)
100.88
Atom Properties and SDF files¶
Note This section describes functionality added in the 2019.03.1 release of the RDKit.
By default the rdkit.Chem.rdmolfiles.SDMolSupplier and rdkit.Chem.rdmolfiles.ForwardSDMolSupplier classes
(RDKit::SDMolSupplier and RDKit::ForwardMolSupplier in C++) can now recognize some molecular properties as property lists
and them into atomic properties. Properties with names that start with atom.prop, atom.iprop, atom.dprop, or atom.bprop
are converted to atomic properties of type string, int (64 bit), double, or bool respectively.
Here’s a sample block from an SDF that demonstrates all of the features, they are explained below:
property_example
RDKit 2D
3 3 0 0 0 0 0 0 0 0999 V2000
0.8660 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.4330 0.7500 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
-0.4330 -0.7500 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
2 3 1 0
3 1 1 0
M END
> <atom.dprop.PartialCharge> (1)
0.008 -0.314 0.008
> <atom.iprop.NumHeavyNeighbors> (1)
2 2 2
> <atom.prop.AtomLabel> (1)
C1 N2 C3
> <atom.bprop.IsCarbon> (1)
1 0 1
> <atom.prop.PartiallyMissing> (1)
one n/a three
> <atom.iprop.PartiallyMissingInt> (1)
[?] 2 2 ?
$$$$
Every atom property list should contain a number of space-delimited elements equal to the number of atoms.
Missing values are, by default, indicated with the string n/a. The missing value marker can be changed by beginning
the property list with a value in square brackets. So, for example, the property PartiallyMissing is set to “one”
for atom 0, “three” for atom 2, and is not set for atom 1. Similarly the property PartiallyMissingInt is set to 2 for atom 0, 2 for atom 1,
and is not set for atom 2.
This behavior is enabled by default and can be turned on/off with the
rdkit.Chem.rdmolfiles.SetProcessPropertyLists method.
If you have atom properties that you would like to have written to SDF files, you can use the functions
rdkit.Chem.rdmolfiles.CreateAtomStringPropertyList(), rdkit.Chem.rdmolfiles.CreateAtomIntPropertyList(),
rdkit.Chem.rdmolfiles.CreateAtomDoublePropertyList(), or rdkit.Chem.rdmolfiles.CreateAtomBoolPropertyList() :
>>> m = Chem.MolFromSmiles('CO')
>>> m.GetAtomWithIdx(0).SetDoubleProp('foo',3.14)
>>> Chem.CreateAtomDoublePropertyList(m,'foo')
>>> m.GetProp('atom.dprop.foo')
'3.1400000000000001 n/a'
>>> from io import StringIO
>>> sio = StringIO()
>>> w = Chem.SDWriter(sio)
>>> w.write(m)
>>> w=None
>>> print(sio.getvalue())
RDKit 2D
2 1 0 0 0 0 0 0 0 0999 V2000
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2990 0.7500 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
M END
> <atom.dprop.foo> (1)
3.1400000000000001 n/a
$$$$
Support for Enhanced Stereochemistry¶
Overview¶
We are going to follow, at least for the initial implementation, the enhanced stereo representation
used in V3k mol files: groups of atoms with specified stereochemistry with an ABS, AND, or OR
marker indicating what is known. The general idea is that AND indicates mixtures and OR indicates unknown single substances.
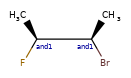
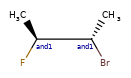
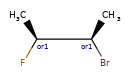
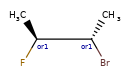
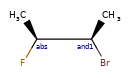
Here are some illustrations of what the various combinations mean:
| What’s drawn | Mixture? | What it means |
|---|---|---|
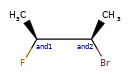 |
mixture | 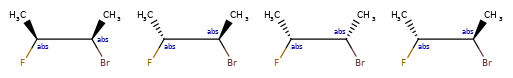 |
 |
mixture | 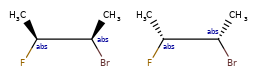 |
 |
mixture | 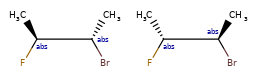 |
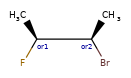 |
single | |
 |
single | |
 |
single | |
 |
mixture | 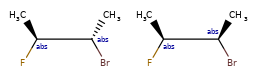 |
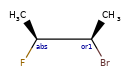 |
single | |
Representation¶
Stored as a vector of rdkit.Chem.rdchem.StereoGroup objects on a molecule. Each StereoGroup keeps track of its type
and the set of atoms that make it up.
Use cases¶
The initial target is to not lose data on an V3k mol -> RDKit -> V3k mol round trip. Manipulation, depiction, and searching are future goals.
It is possible to enumerate the elements of a StereoGroup using the function rdkit.Chem.EnumerateStereoisomers.EumerateStereoisomers(), which also
preserves membership in the original StereoGroup.
>>> m = Chem.MolFromSmiles('C[C@H](F)C[C@H](O)Cl |&1:1|')
>>> m.GetStereoGroups()[0].GetGroupType()
rdkit.Chem.rdchem.StereoGroupType.STEREO_AND
>>> [x.GetIdx() for x in m.GetStereoGroups()[0].GetAtoms()]
[1]
>>> from rdkit.Chem.EnumerateStereoisomers import EnumerateStereoisomers
>>> [Chem.MolToCXSmiles(x) for x in EnumerateStereoisomers(m)]
['C[C@@H](F)C[C@H](O)Cl |&1:1|', 'C[C@H](F)C[C@H](O)Cl |&1:1|']
Reactions also preserve StereoGroup``s. Product atoms are included in the ``StereoGroup as long as the reaction doesn’t create or destroy chirality at the atom.
>>> def clearAllAtomProps(mol):
... """So that atom mapping isn't shown"""
... for atom in mol.GetAtoms():
... for key in atom.GetPropsAsDict():
... atom.ClearProp(key)
...
>>> rxn = AllChem.ReactionFromSmarts('[C:1]F >> [C:1]Br')
>>> ps=rxn.RunReactants([m])
>>> clearAllAtomProps(ps[0][0])
>>> Chem.MolToCXSmiles(ps[0][0])
'C[C@H](Br)C[C@H](O)Cl |&1:1|'
Footnotes
| [1] | http://www.daylight.com/dayhtml/doc/theory/theory.smirks.html |
| [2] | (1, 2) http://www.daylight.com/dayhtml/doc/theory/theory.smiles.html |
| [3] | (1, 2, 3) http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html |
| [4] | https://docs.chemaxon.com/display/docs/ChemAxon+Extended+SMILES+and+SMARTS+-+CXSMILES+and+CXSMARTS |
| [5] | Thanks to James Davidson for this example. |
| [6] | Thanks to JP Ebejer and Paul Finn for this example. |
License¶

This document is copyright (C) 2007-2019 by Greg Landrum
This work is licensed under the Creative Commons Attribution-ShareAlike 4.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by-sa/4.0/ or send a letter to Creative Commons, 543 Howard Street, 5th Floor, San Francisco, California, 94105, USA.
The intent of this license is similar to that of the RDKit itself. In simple words: “Do whatever you want with it, but please give us some credit.”